Research

Interaction of synthetic nanomaterials with biological membranesThe last two decades have been characterized by an extraordinary growth in the production and commercialization of nano-sized materials. Nanometer size materials offer unprecedented opportunities in many fields of technology, including pharmaceutical and medical technology. At the same time, safety is a major concern when new materials are produced, and especially so for nanomaterials, since their properties are very different from the properties of bulk materials with the same chemical composition, and are difficult to predict. The increasing use of synthetic nanoparticles makes it important to understand how such particles interact with biological matter and particularly with biological membranes, which are the first barrier encountered by any particle entering an organism. Our goal is to understand different aspects of the interaction between synthetic nanomaterials and biological membranes at the molecular level, using computer simulations. We focus especially on pure carbon nanoparticles, such as fullerenes and carbon nanotubes, and on industrial polymers and the additives therein. Simulations are carried out at the coarse-grained leven and at the all-atom level, depending on the specific problem. |
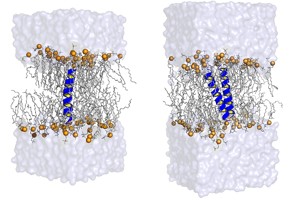
Protein-lipid interactions and lipid-mediated interactions between membrane proteinsCollaboration: Peter Tieleman (U. Calgary, Canada)
Lipid membranes play a major role idetermining membrane protein structure and
functioning. The structure of membrane proteins is only stable within specific membranes,
which makes it difficult to crystallize membrane proteins in detergents.
The orientation of membrane proteins
depends on the mismatch between the length of the hydrophobic part
of the peptide and the hydrophobic thickness of the lipid bilayer.
Conformational equilibria, and therefore functioning of membrane proteins, depend
on membrane properties, such as electrostatic properties, structural properties, elastic properties,
and even specific protein-lipid interactions.
Our goal is to understand how lipid membranes affect
membrane protein structure and functions, as well as the interaction between multiple proteins.
We focus particularly on non-specific effects, which do not depend on specific lipid-protein interactions
and are well captured by coarse-grained molecular models.
|
MARTINI force field developmentCollaborations: S.J. Marrink (U. Groningen, the Netherlands), G. Rossi (U. Genoa, Italy)
A large number of biologically relevant motions involve relatively large systems,
with lateral size of tens of nanometers, and occur on time scales of tens of microseconds.
Those length and time scales are currently difficult to reach in molecular simulations at the atomistic level.
Coarse-grained descriptions of the systems allow faster simulations
and still retain many important properties of molecular systems.
|
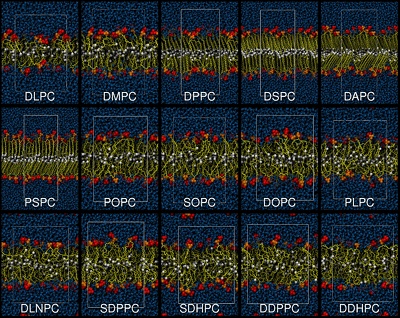
Development of a united-atoms force field for lipidsCollaborations: D. Peter Tieleman (U. Calgary, Canada), Emppu Salonen (Aalto U., Finland)
A number of limitations affect current simulations of lipid aggregates. These limitations
are due to the quality of present force fields and to the methodology used,
which is not ideal in the case of systems with interfaces.
I started working on a new lipid force field already in 2005, together with Prof Tieleman, and
the development is still ongoing.
|